


N-Fluorobenzenesulfonimide used as fluorine source
Jan 22,2024
Description
In 1991, N-fluorobenzenesulfonimide was synthesized by Differding and co-workers. It was prepared from N-(phenylsulfonyl)benzenesulfonamide in good yield by reacting with 10% F2/N2 in acetonitrile at −40 °C. NFSI is a stable and non-hygroscopic crystalline solid with a 114–116 °C mp.
N-Fluorobenzenesulfonimide (NFSI) is one of the most popular commercially available N–F reagents. This mild, stable, and highly soluble crystalline solid was not only utilized as fluorination but also as an amination reagent in various transformations of aliphatic and aromatic compounds. NFSI was shown to fluorinate a variety of nucleophiles. Trimethylsilyl enol ethers, enolate anions of ketones and esters, and aryl- and vinyl lithiums were fluorinated with NFSI in moderate to high yields. Although NFSI could also fluorinate aromatics such as anisole, toluene, and acetanilide, these reactions required neat conditions and high temperatures, indicating that NFSI was not so powerful[1].

Transition-metal-catalyzed reactions
In 2013, Xu and co-workers disclosed for the first time the usefulness of palladium catalysts for the regioselective C–H bond fluorination of (hetero)aromatic compounds with NFSI[2]. Thus, in the presence of Pd(OAc)2/TFA (trifluoroacetic acid) combination as a catalytic system in a mixed solvent of MeNO2 and MeCN (with different ratios) at 110 °C, fluorination of (hetero)arenes 1 that bear N-heterocyclic directing groups (e.g., quinoxaline, pyrazole, benzo[d]oxazole, and pyrazine) with NFSI furnished the corresponding ortho-mono fluorinated products 2 in moderate to good yields. The results demonstrated that the substrates bearing electron-donating groups were relatively more favorable in this transformation, and the relative activation rates of directing groups followed the order quinoxaline ≈ pyrazole > pyrazine > benzo[d]oxazole.
In 2005, Yamamoto and co-workers demonstrated an efficient methodology for directed C–H fluorination of (hetero)arenes 7 under zirconium-catalyzed conditions. NFSI was employed as the fluorine source, and ZrCl4 was found to be the most effective Lewis acid catalyst. The reactions were done in DCM at room temperature without consuming additional chemicals and provided the corresponding mono-fluorinated products 8 in moderate to good yields. However, only three examples were demonstrated for such a fluorination process. The protocol was also compatible with chlorination, bromination, and iodination reactions when NCS, NBS, and NIS were used.
In 2017, Ding, Zhang, and Li demonstrated interesting Ni-catalyzed fluorination of 8-amido quinolines in a highly regioselective manner at the C5 position by using NFSI as the oxidant and fluorine source. The method only made use of NiSO4, and neither additive nor ligand was required for this transformation. Various quinolines 9 containing aromatic, heteroaromatic, aliphatic, long-chain, and sterically bulky amides were applied to afford the corresponding 5-fluorinated quinolines 10 with yields ranging from 32% to 46%. The results revealed that the presence of free N–H of the amide and the N atom in the quinoline skeleton was indispensable for this transformation. The authors showed that the amido groups could be efficiently converted to the free amino (–NH2) group by hydrolysis under alkaline conditions without damaging the C–F bond. The control experiments testified that this fluorination reaction underwent a free radical cross-coupling pathway.
Transition-metal-free reactions
After pioneering works by Kirk and colleagues on tBuLi-mediated regioselective C4-fluorination of (5-methoxy-indol-3-yl)-N, N-dimethylmethanamine with NFSI and O'Neill's research group on catalyst-free C5-fluorination of 6-methoxy-8-nitroquinoline, the first general report of the transition-metal-free fluorination of aromatic C–H bonds using NFSI as a fluorinating reagent was published in 2009 by Andreev et al. They showed that in the absence of any catalyst or additive, the reaction of a variety of electron-rich arenes 11, including anisoles, phenols, toluene, and naphthalenes with NFSI under solvent-free conditions furnished the mono-fluorinated-products 12 in poor to good yields. However, the fluorinations occurred not only on one C–H bond but resulted in mixtures of regioisometric mono-fluorides in some cases, e.g., for m-xylene and 1-naphthol. Moreover, mono- versus di-fluorination selectivity could not be controlled for substrates without ortho and para substituents.
References
[1] Teruo Umemoto, Gerald B Hammond, Yuhao Yang. “Development of N-F fluorinating agents and their fluorinations: Historical perspective.” Beilstein Journal of Organic Chemistry (2021): 1752–1813.
[2] Gu, Qiang and Esmail Vessally. “N-Fluorobenzenesulfonimide: a useful and versatile reagent for the direct fluorination and amination of (hetero)aromatic C–H bonds.” RSC Advances 28 (2020): 16756–16768.
- Related articles
- Related Qustion
- The role of N-fluorobisbenzenesulfonamide in chemical synthesis Mar 31, 2022
N-Fluorobenzenesulfonimide (NFSI) is a colorless crystalline powder with a melting point of 114–116 °C

Tert-butyl acetate is a readily available low-cost solvent produced industrially from acetic acid and isobutylene. Its good safety profile makes it suitable for widespread use in catalytic direct amidation reactions.....
Jan 22,2024Chemical Reagents?Pexidartinib (PLX3397) is a novel, orally available small molecule tyrosine kinase inhibitor (TKI) with potent and selective activity against the colony-stimulating factor 1 (CSF1) receptor.....
Jan 22,2024Inhibitors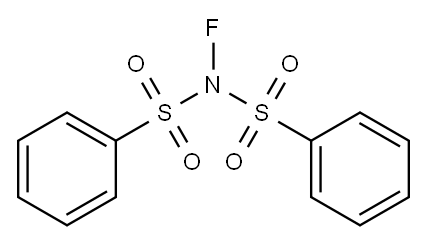
N-Fluorobenzenesulfonimide
133745-75-2You may like
N-Fluorobenzenesulfonimide manufacturers
- N-Fluorobenzenesulfonimide
-

- $18.00 / 10kg
- 2024-05-10
- CAS:133745-75-2
- Min. Order: 1kg
- Purity: 99.9
- Supply Ability: 5000
- N-Fluorobenzenesulfonimide
-

- $30.00/ kg
- 2024-04-28
- CAS:133745-75-2
- Min. Order: 1kg
- Purity: 99%
- Supply Ability: 5000kg/Month
- N-fluorobenzenesulfonamide
-

- $15.00/ kg
- 2024-04-23
- CAS:133745-75-2
- Min. Order: 1kg
- Purity: 99.912%
- Supply Ability: 10ton